The Office of Research at the University of Michigan Medical School is constantly striving to enhance the research enterprise, including maintaining an investigator-focused infrastructure, facilitating and diversifying investigators' avenues for funding, and streamlining research processes.


Whether you've got a question about the next "station" or have hit a break in the tracks, the Medical School Office of Research is committed to providing personal service that helps you navigate your own project route on a path to successful research.

What are bold ideas? From diverse disciplines across the Medical School, Research Scouts were given $150,000 and empowered and motivated to support their fellow researchers’ bold ideas.
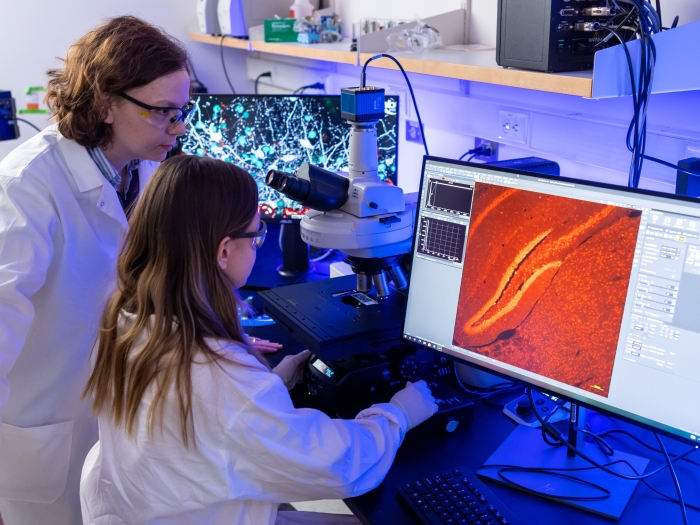
The University of Michigan Medical School’s strategic research plan, “Great Minds, Greater Discoveries,” is focused largely on our people, who remain our greatest resource.

Find funding opportunities on Competition Space, an innovative online platform that streamlines the process of finding, and applying for, funding opportunities through the U-M Medical School.
Below are a list of quick links to our most-visited services. We look forward to working with you!

1301 Catherine Street
Ann Arbor, MI 48109-5624
North Campus Research Complex (NCRC)
2800 Plymouth Road Building 520, 3rd Floor
Ann Arbor, MI 48109-2800






At Michigan Medicine, we unequivocally recognize racism as a public health issue and we stand together against bias and inequality.